Concepts of biosimilars
Biosimilar development, under EMA and FDA regulations, focuses on the totality of the evidence of an all-encompassing comparability exercise. In order to receive the biosimilar regulatory designation, the proposed product must pass non-clinical analytical, pharmacological as well as confirmatory clinical studies to remove in a step-wise approach the uncertainty of comparability and subsequently to demonstrate biosimilarity to a reference product. The development of a biosimilar can take 8–10 years at an estimated cost of $100–200 million (in contrast, these values for a generic drug are 3-5 years and $1-5 million, respectively).1-3
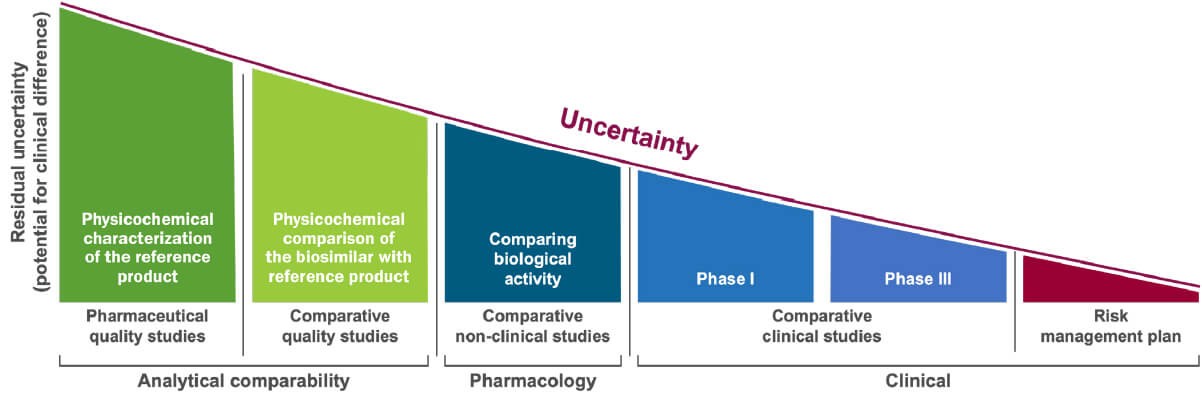
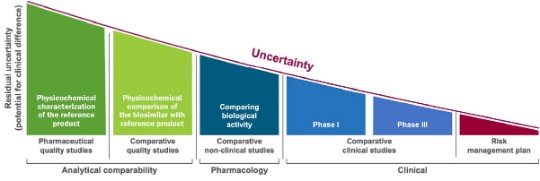
Biosimilar development steps (please click on different steps for more explanation). Adapted from Wolff-Holz E, et al. ESMO Open 2018;3(6):e000420; Mihalcik L, et al. Regul Toxicol Pharmacol 2021;122:104912.
Biosimilar products are modeled after a product with an established risk-benefit profile, thoroughly studied in large nonclinical and clinical study programs. There is usually a wealth of information available for these reference products, including years of commercial use in large and diverse patient populations.2,3
The focus of biosimilar development is not to establish a risk-benefit profile, but to demonstrate similarity to a reference product. This starts with thorough biochemical characterization of both the proposed biosimilar and the reference product.2,3
Healthcare authorities require that the biochemical fingerprint of critical and non-critical product quality attributes (CQA) must be defined for each biologic with standardized, so called “state of the art” analytical methods and replicated in a biosimilar. The extent of variation in each of the CQA must be characterized for the originator molecule and systematically matched as closely as possible, ensuring that the biosimilar is reverse engineered to meet similar specifications.2,4,5
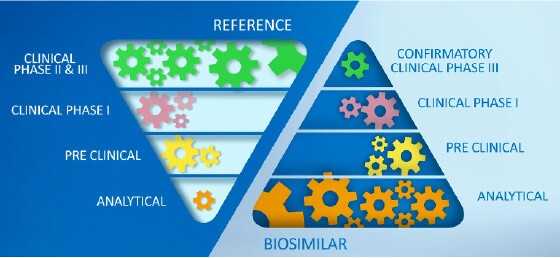
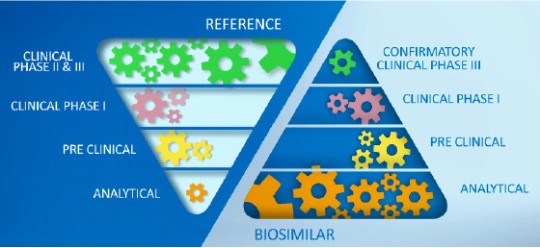
In contrast to a novel therapy, in biosimilar development there is a greater emphasis on the analytical characterization of both, the proposed biosimilar and the reference product. Adapted from Wolff-Holz E, et al.4
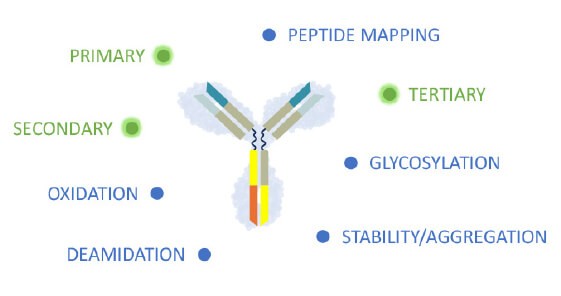
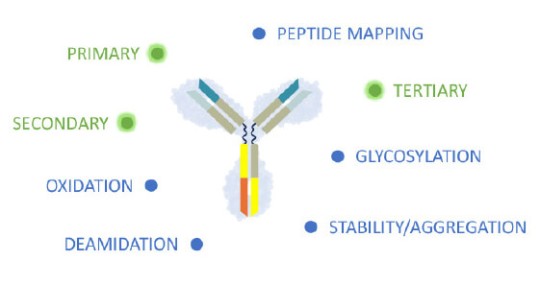
Primary, secondary and higher structure define the biochemical fingerprint and CQA of the recombinant protein.
Replicating highly complex biologics has been made possible due to enormous analytical advances, reducing the need for extensive clinical evaluation. Therefore, the clinical program of a biosimilar aims to confirm that proteins with highly similar structures and biochemical pactivity have similar pharmacokinetic (PK) and pharmacodynamic (PD) profiles.2
The phase I and phase III clinical studies are designed to demonstrate equivalence in PK parameters and in efficacy endpoints and to compare safety and immunogenicity profiles between a biosimilar and its reference product. A phase 2 dose finding study is not required as the same, well-established dosing is used for the biosimilar and the reference product. An equivalence study design is used which aims to prove that differences in response between the biosimilar and reference product are clinically unimportant – i.e. neither the biosimilar nor the originator is superior or inferior to the other. Endpoints and equivalence margins are predefined and endorsed by the regulators scientific advice.5
Similar to the reference product, ongoing pharmacovigilance activities will further evaluate effectiveness and safety profiles of biosimilars post approval.2,3
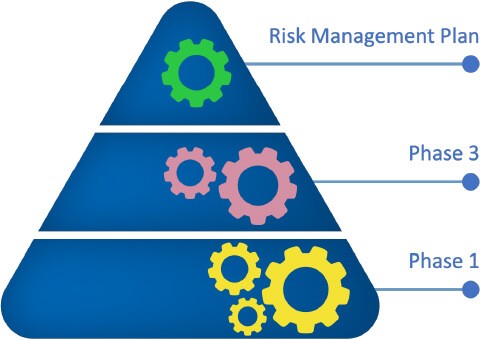
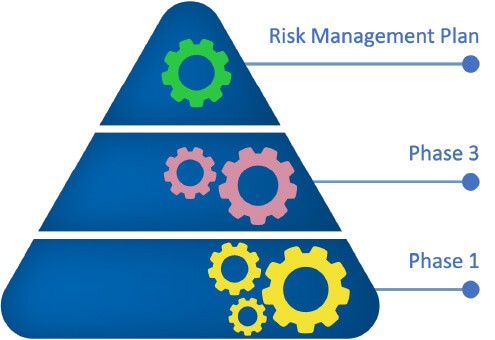
Pharmacovigilance surveillance and a risk management plan for identified and possible risks, and with immunogenicity being given particular attention to, are measures to further support the safe use of biosimilars post approval.1-3
The phase 3 study aims to demonstrate equivalence in efficacy endpoints as well to compare efficacy, safety and immunogenicity profiles between a biosimilar and its reference product. The study population should be representative of approved therapeutic indication(s) of the reference product and be sensitive for detecting potential differences between the biosimilar and the reference. A dose finding phase 2 study is not required.1,3
The phase 1 clinical study aims to demonstrate equivalence in PK and comparable PD of the biosimilar and its reference product. The design of the PK study depends on a number of factors, including clinical context, safety profile and the PK of the reference agent.1,3
Extrapolation
When similarity of a biosimilar to a reference biologic has been comprehensively shown in a key indication, extrapolation of efficacy and safety data to other indication(s) of the reference product may be possible. The European regulatory agency (EMA) and the US FDA require an acceptable scientific justification to extrapolate clinical data from one indication to another.7,8
Requirements of reasonable scientific justification for extrapolation include: 1) the principle of similar mechanism of action by understanding the mode of action of the reference product in different indications, 2) similar pharmacokinetic profiles across indications, 3) using a sensitive patient population for clinical studies to detect clinically meaningful differences in efficacy and safety profiles, in particular excluding risk of increased immunogenicity.7,8


FDA. Biosimilar product Regulatory Review and Approval. Available from: https://www.fda.gov/files/drugs/published/Biosimilar-Product-Regulatory-Review-and-Approval.pdf, accessed Feb 2025.
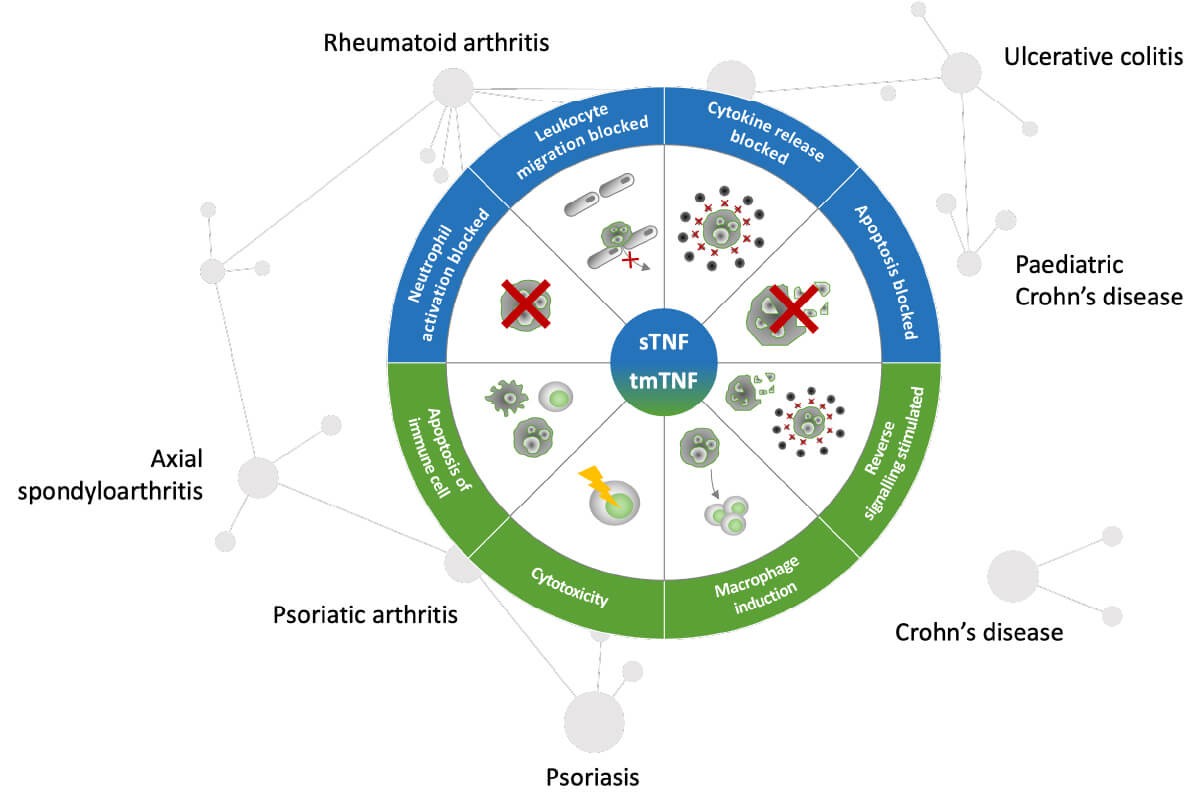
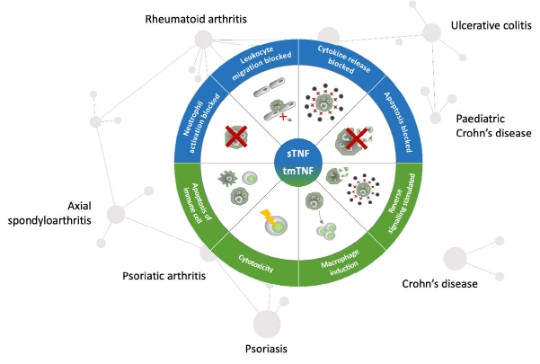
Pharmacological determinants of anti-TNFs in chronic immune mediated inflammatory diseases.
As an example, the pharmacological activity of TNF alpha inhibition with anti-TNFs is well established for different indications of chronic immune mediated inflammatory diseases. To date there are 17 anti-TNF biosimilars approved by the European Medicines Agency. While most confirmatory phase 3 equivalence studies of biosimilars were performed in RA as the sensitive patient population, they are widely used across different indications.9,10
Post-marketing surveillance shows that until August 2020, the highest patient exposure of biosimilars in Europe was with anti-TNFs. There were no biosimilar-specific adverse effects, and no product was withdrawn for safety reasons.10
Interchangeability
Interchangeability refers to exchanging one medicine for another that is expected to achieve the same clinical efficacy with the same/a comparable safety profile. For biosimilars this means that switching between a biosimilar and its licensed reference product or another biosimilar can be done without concerns of efficacy, tolerability or immunogenicity.11
In clinical practice, the replacing of a reference product with a biosimilar, a biosimilar with a reference product, or one biosimilar with another is known as switching, if mandated by the physician. In contrast, the practice of dispensing one medicine instead of another medicine without consulting the prescriber refers to auto-substitution.11
From a regulatory viewpoint, the definitions and legal status of the term interchangeability vary across the jurisdictions of EMA and US FDA:
For the EMA, the term interchangeability is a scientific rationale and should not be equated to the term auto-substitution. It acknowledges that interchangeable use of biosimilars is already practiced in many European Member States. In September 2022, the EMA released a joint position statement to harmonize the EU approach with the aim to bring more clarity for healthcare providers and to help more patients to have access to biological medicines across the EU.12
The EMA considers that once a biosimilar is approved in the EU, it is interchangeable, which means the biosimilar can be used instead of its reference product (or vice versa) or one biosimilar can be replaced with another biosimilar of the same reference product without patients experiencing any changes in the clinical effect. No additional systematic switch studies are needed to support the interchangeability. Decisions regarding substitution (the practice of dispensing one medicine instead of another medicine without consulting the prescriber), are not within the remit of the EMA and are managed by individual European member states.12


By the US FDA, the term interchangeability is a regulatory designation. It is the legal justification that a reference product can be substituted with an interchangeable biosimilar at the pharmacy without the intervention of the prescribing healthcare professional.11,13 It only permits a biosimilar to be automatically substituted for its licensed reference product, not other biosimilars of the same reference product.14,15
In 2019, the FDA released guidance to industry on considerations to demonstrate interchangeability of a biosimilar with its reference product. At that time, the FDA had a very strong position that data from “switching studies” must be required to assess the risk, in terms of safety and diminished efficacy when switching between reference product and the biosimilar product. Since then, the scientific approach to when a switching study, or studies, may be needed to support a demonstration of interchangeability has evolved. The FDA acknowledges that 10 years valuable experience, reviewing both biosimilar and interchangeable biosimilar medications, showed that biosimilars and interchangeable biosimilars meet the same high standard of biosimilarity for FDA approval and both are as safe and effective as the reference product. Hence, the FDA is in the process of updating its guidance to the pharmaceutical industry on interchangeable biosimilars, with recommendation of fewer tests to demonstrate interchangeability. The US regulatory agency also recently released a list of “9 Things to Know About Biosimilars and Interchangeable Biosimilars”.13,14,16


References
1.Federal Trade Commission. Emerging Health Care Issues: Follow-on Biologic Drug Competition. Available at: https://www.ftc.gov/sites/default/files/documents/reports/emerging-health-care-issues-follow-biologic-drug-competition-federal-trade-commission-report/p083901biologicsreport.pdf, accessed December 2024.
2.Vulto AG & Jaquez OA. Rheumatology (Oxford) 2017; 56(Suppl 4):iv14–iv29.
3.Declerck P & Rezk MF. Rheumatology (Oxford) 2017; 56(suppl 4):iv4– iv13.
4.Wolff-Holz E, et al. ESMO Open 2018;3(6):e000420.
5.Mascarenhas-Melo F, et al. Pharmaceuticals (Basel). 2024 Feb; 17(2):235.
6.Vandekerckhove K, et al. AAPS J. 2018:20(4):68.
7.Weise M, et al. Blood 2014;124(22):3191–3196.
8.Weise M, et al. Blood 2012;120(26):5111-5117.
9.EMA. Medicines for Human Use. Available from: https://www.ema.europa.eu/en/search?search_api_fulltext=biosimilars&f%5B0%5D=ema_search_categories%3A83&f%5B1%5D=ema_medicine_bundle%3Aema_medicine&f%5B2%5D=ema_med_status%3Aauthorised&landing_from=73303
10.Kurki P, et al. Drugs. 2021;81(16):1881-1896.
11.Kurki P, et al. BioDrugs 2017;31(2):83-91.
12.EMA – HMA. Biosimilar‘s interchangeability – Joint statement. 2022, April 2023. Available from: https://www.ema.europa.eu/en/documents/public-statement/statement-scientific-rationale-supporting-interchangeability-biosimilar-medicines-eu_en.pdf, accessed December 2024.
13.FDA. Implementation of the Biologics Price Competition and Innovation Act. 2019. Available from: https://www.fda.gov/drugs/guidance-compliance-regulatory-information/implementation-biologics-price-competition-and-innovation-act-2009, accessed December 2024.
14.FDA. 9 Things to Know About Biosimilars and Interchangeable Biosimilars. Available from: https://www.fda.gov/drugs/things-know-about/9-things-know-about-biosimilars-and-interchangeable-biosimilars, accessed December 2024.
15.Afzali A, et al. Adv Ther. 2021;38(5):2077-2093.
16.FDA. Considerations in Demonstrating Interchangeability With a Reference Product Guidance for Industry. Available from: https://www.fda.gov/media/124907/download, accessed December 2024.
Abbreviations:
CQAs Critical Quality Attributes
EMA European Medicines Agency
FDA Food and Drug Administration (USA)
MoA Mechanism of action
PD Pharmacodynamics
PK Pharmacokinetics
Learn more
Read more about biosimilars concepts in the publications:
Vulto AG and Jaquez A, 2017 Rheumatology (Oxford) 1;56
The process defines the product: what really matters in biosimilar design and production
Biologic drugs are highly complex molecules and their key characteristics (known as critical quality attributes or CQAs) can vary based on post-translational modifications that occur. The extent of variation in each CQA must be matched by biosimilar developers to the originator molecule to ensure biosimilarity. As analytical tools that measure differences at the molecular level are more sensitive and specific than tools that measure clinical differences, for example in clinical trials, biosimilar development has a greater focus on preclinical attributes, with phase 3 trials (if needed) being confirmatory. A well-controlled manufacturing process ensures that biosimilars consistently match the fingerprint of the originator molecule.
Declerck P, Rezk MF, Rheumatology (Oxford) 2017 Aug 1;56(suppl_4):iv4-iv13.
The road from development to approval: evaluating the body of evidence to confirm biosimilarity
Similarity of biosimilars to originator molecules is established through a step-wise, comprehensive comparability exercise. This includes assessment of physicochemical, biological and immunochemical properties, in vivo pharmacology studies, phase 1 clinical studies (if appropriate) to determine pharmacokinetic and pharmacodynamic characteristics, and phase 3 clinical studies to determine efficacy, safety profile and tolerability. Post-approval risk management is required and includes implementation of pharmacovigilance systems. The robust nature of this biosimilarity exercise is an important element when considering these agents.
Vanderkerckhove K., Seidl A. et al,. 2018, AAPS J. 2018: 20:68
Rational selection, criticality assessment, and tiering of quality attributes and test methods for analytical similarity evaluation of biosimilars
Regulatory agencies recommend biosimilars are assessed against their originator molecules in a stepwise fashion, starting with a detailed structural and functional analysis, followed by in vivo studies. If evidence from these analyses demonstrate similarity, this may justify a targeted clinical development plan. Therefore, careful design of the analytical phase of the process is critical.
Weise M, Kurki P, et al. Blood 2014;124:3191–6.
Biosimilars: the science of extrapolation
This article addresses the concerns frequently raised in the medical community regarding the use of biosimilars in extrapolated indications and explains the underlying scientific and regulatory decision making, including some real-life examples from recently licensed biosimilars. This article is an extension to a paper published previously by Weise et al. (Blood 2012;120:5111–5117) which explained the principles of biosimilar development in general.
Kurki P, Barry S, et al. Drugs. 2021;81(16):1881-1896.
Safety, Immunogenicity and Interchangeability of Biosimilar Monoclonal Antibodies and Fusion Proteins: A Regulatory Perspective
This is a comprehensive analysis of prelicensing and postmarketing safety surveillance reports from the European Medicines Agency (EMA) of all biosimilar mAbs and fusion proteins approved before August 2020. The findings support the conclusion that EU-approved biosimilars are highly similar and interchangeable with their reference products, negating the need for additional systematic switch studies.
Ebbers HC, Schellekens H. Drug Discov Today. 2019 Oct;24(10):1963-1967.
Are we ready to close the discussion on the interchangeability of biosimilars?
Discussion about the interchangeability of biosimilars, where one medicine can be substituted for another without clinical consequence, persists. Biosimilars are approved based on a rigorous, step-wise comparability exercise demonstrating no clinically meaningful differences compared to the originator products. Furthermore, there are no data to suggest that the risk of immunogenicity is greater when switching to a biosimilar than when switching between different batches of the same originator biologic. The nocebo effect reinforces the need for physician involvement when switching but is not limited to biosimilars. For biosimilars to deliver on their promise, patients and physicians should be confident that they can be safely and effectively switched.
Learn through a series of animated videos the essential facts about the science behind the development of biosimilars:









